
NORTH CHICAGO, Ill., July 29, 2022 /PRNewswire/ — AbbVie a reçu aujourd’hui l’approbation européenne pour son traitement oral upadacitinib (RINVOQ). Parmi ses nombreuses utilisations, il est désormais approuvé pour le traitement de la spondyloarthrite axiale active non radiographique (nr-axSpA) chez les patients adultes, comme indiqué par des niveaux élevés de CRP et/ou une IRM anormale, les patients souffrant de douleurs qui ont répondu de manière inadéquate aux AINS.
Pendant des années, les prestataires de soins et les patients ont eu des options de traitement limitées pour gérer la spondyloarthrite axiale, qui peut provoquer des douleurs dorsales, une raideur et des dommages irréversibles à la colonne vertébrale. AbbVie est fière d’offrir RINVOQ comme première option thérapeutique de sa catégorie, maintenant approuvée dans l’Union européenne pour les adultes vivant avec une spondylarthrite nr-axiale présentant des signes objectifs d’inflammation et une réponse inadéquate aux AINS. RINVOQ est le premier et le seul inhibiteur des JAK approuvé pour le traitement des patients atteints de spondylarthrite axiale, qui comprend le nr-axSpA et la spondylarthrite ankylosante.
Thomas Hudson, M.D., vice-président senior de la recherche et du développement, responsable scientifique, AbbVie.
La spondyloarthrite axiale est une maladie chronique qui implique une inflammation des articulations et peut entraîner des douleurs dorsales sévères.3,4,5 Il existe deux types d’AxSpA qui ont été définis cliniquement comme ankylosant et non ankylosant ; tous deux impliquent une inflammation des articulations et peuvent entraîner des douleurs dorsales sévères, La polyarthrite rhumatoïde (PR) est une maladie inflammatoire chronique qui affecte principalement les mains et les pieds. La PR peut également affecter d’autres articulations, notamment celles du bassin. L’un de ces autres types, appelé spondyloarthrite axiale non radiographique (nr-axSpA), se développe généralement sur une période de 2 à 10 ans à partir de.
“L’approbation par la Commission européenne du RINVOQ pour le traitement de la spondylarthrite nr-axiale offre aux médecins de l’Union européenne une nouvelle option thérapeutique importante dont l’efficacité a été prouvée dans les populations de patients atteints de spondylarthrite nr-axiale et de spondylarthrite ankylosante”, a déclaré Filip Van den Bosch, M.D.,** investigateur de SELECT-AXIS 2 et professeur au département de rhumatologie de l’hôpital universitaire de Gand. “Vivre avec le nr-axSpA peut poser de nombreux défis et avoir un impact significatif sur la qualité de vie du patient. Une prise en charge précoce et efficace de la maladie chez les patients atteints de nr-axSpA actif est essentielle pour améliorer les résultats de santé.”
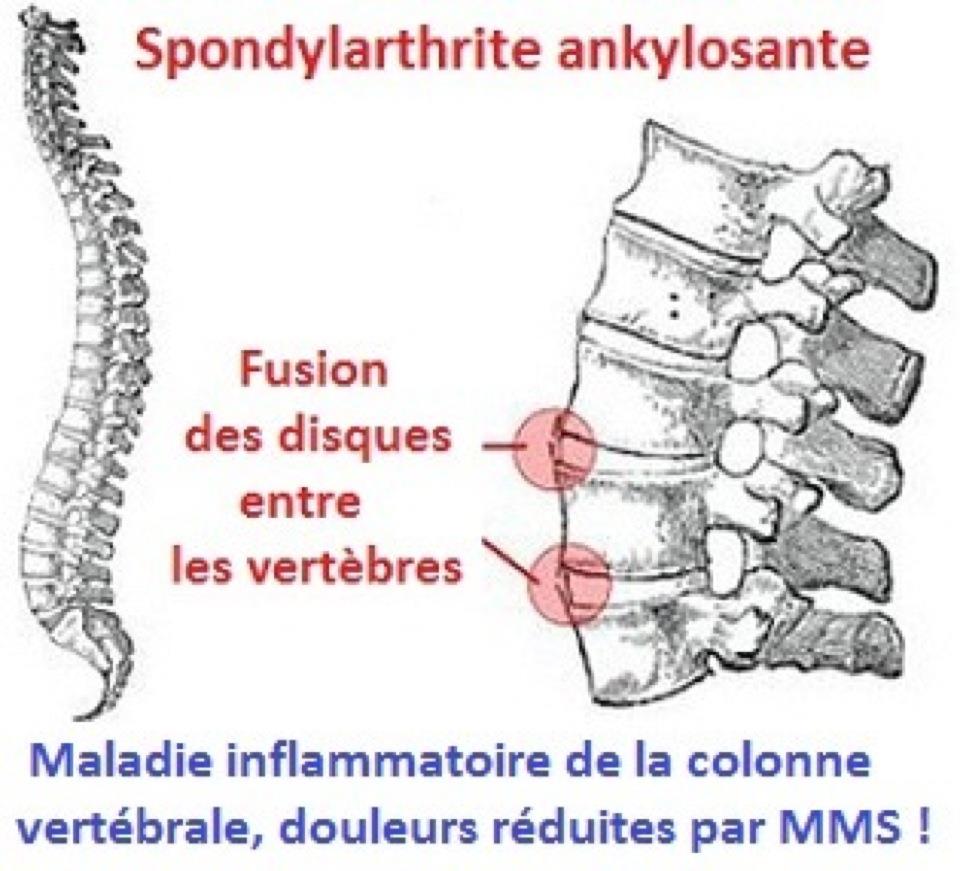
AbbVie a précédemment divulgué les premiers résultats de l’étude clinique de phase 3 SELECT-AXIS 2 sur le nr-axSpA et les résultats complets ont été publiés dans The Lancet. Les résultats de cette étude montrent qu’une proportion significativement plus importante de patients ayant reçu RINVOQ 15 mg ont atteint l’ASAS40 à la semaine 14 (45 %) par rapport à ceux ayant reçu le placebo (23 %). Cette différence a été statistiquement significative. Les données montrent des améliorations significatives dans 12 des 14 critères d’évaluation secondaires par rapport au placebo. Aucun nouveau risque associé à ce médicament n’est également apparu au cours de la période d’étude. D’après les données recueillies jusqu’à la semaine 14, la fréquence des événements indésirables était similaire chez les patients prenant RINVOQ et ceux recevant le placebo (RINVOQ à 48 % et placebo à 46 %).
L’autorisation de mise sur le marché européenne de nr-axSpA signifie que le médicament est approuvé dans tous les États membres de l’UE, ainsi qu’en Islande, au Liechtenstein, en Irlande du Nord et en Norvège.
Le RINVOQ a été approuvé dans l’UE pour les patients atteints de SA qui ont une réponse inadéquate aux ARMM biologiques, sur la base des résultats de l’essai clinique de phase 3 SELECT-AXIS 2 dans cette population, ainsi que des résultats à deux ans de l’essai clinique de phase 2/3 SELECT-AXIS 1 qui a évalué des patients atteints de SA n’ayant jamais reçu d’ARMM.
AbbVie a récemment dévoilé les résultats de son étude de phase 3 sur les bDMARD de la SA, qui ont révélé qu’une proportion significativement plus importante de patients ayant reçu le RINVOQ 15 mg d’AbbVie ont obtenu une réponse ASAS40 à la semaine 14 (45 % contre 18 %) par rapport au placebo. Les 14 critères d’évaluation secondaires classés ont tous été satisfaits, l’un d’entre eux évaluant les améliorations par rapport aux valeurs initiales de l’activité de la maladie, de la douleur (douleur dorsale totale et nocturne), de la fonction, du score SPARCC à l’IRM (colonne vertébrale), de la mobilité vertébrale, de l’enthésite et de la qualité de vie liée à la santé. Jusqu’à présent, il n’y a pas eu d’effets secondaires graves associés à RINVOQ qui n’étaient pas déjà connus. La majorité des patients qui ont pris le médicament n’ont connu aucun effet indésirable jusqu’à la 14e semaine de traitement.